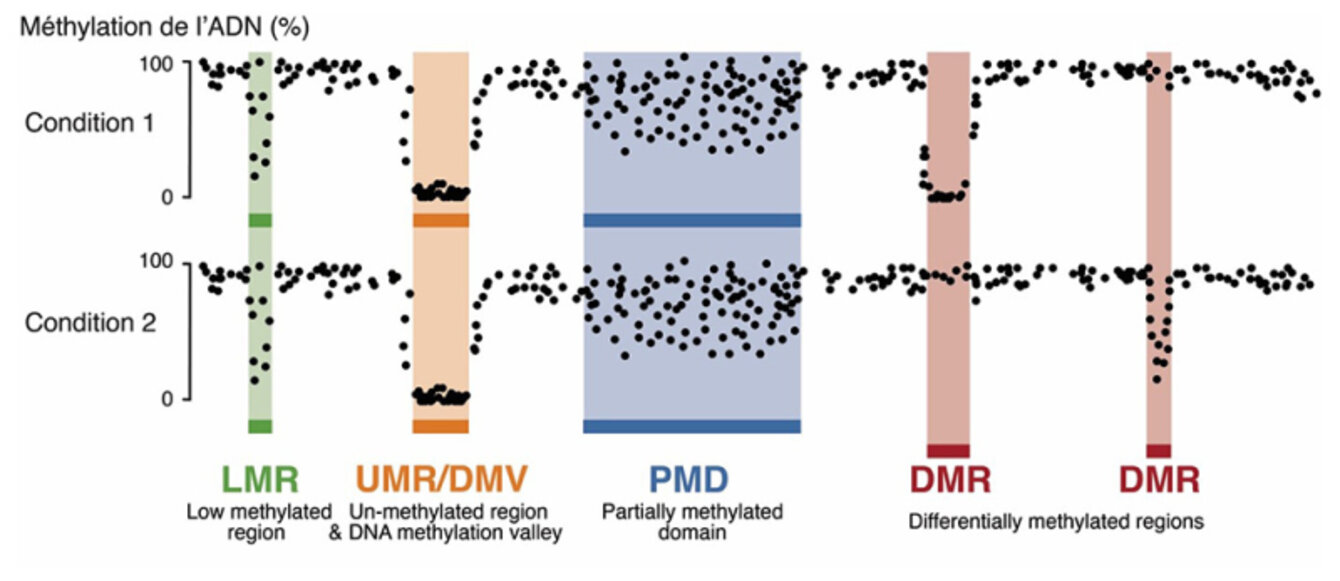
La méthylation de l’ADN est une modification épigénétique permettant de réprimer l’expression des gènes. Dans un article publié dans Nucleic Acids Research, des scientifiques de l'IGBMC et de l'équipe "Régulation épigénétique de l'identité cellulaire" de Michaël Weber décrivent une nouvelle méthode bio-informatique permettant d’analyser les données de méthylation de l’ADN à l’échelle du génome. Cette approche s’avère utile pour mieux comprendre les changements de méthylation de l’ADN induits dans différentes conditions physiologiques ou pathologiques.
Les approches de séquençage à haut débit permettent maintenant de cartographier les niveaux de méthylation de l’ADN sur génome entier. Cependant, le développement de méthodes bio-informatiques est encore nécessaire afin d’analyser les données et d’en extraire le maximum d’information possible.
Les scientifiques ont développé une méthode innovante appelée MethyLasso qui permet une analyse fine des données de méthylation de l’ADN. Cette approche effectue tout d’abord une segmentation des données afin d’identifier des régions du génome qui sont déméthylées et qui correspondent aux régions régulatrices actives liées par des facteurs de transcription. MethyLasso permet aussi d’identifier des régions différentiellement méthylées lors de comparaisons entre deux conditions comme par exemple entre cellules saines et cancéreuses.
Le nouvel outil MethyLasso permet donc aux scientifiques étudiant la méthylation de l’ADN de réaliser une analyse fine de ces données afin de mieux caractériser les dérégulations observées dans différentes conditions pathologiques.
Pour en savoir plus, voir l'actualité publiée sur le site du CNRS Biologie.
Article :
Balaramane D, Spill YG, Weber M*, Bardet AF* (2024). MethyLasso: a segmentation approach to analyze DNA methylation patterns and identify differentially methylated regions from whole-genome datasets. Nucleic Acids Res 18:gkae880. doi: 10.1093/nar/gkae880.